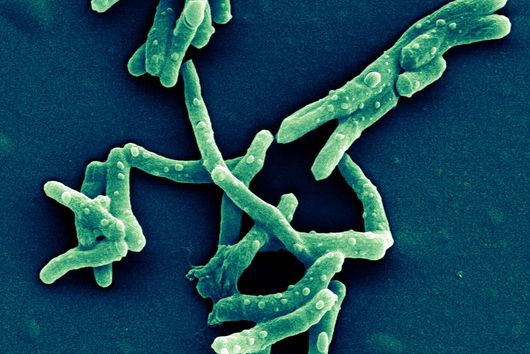
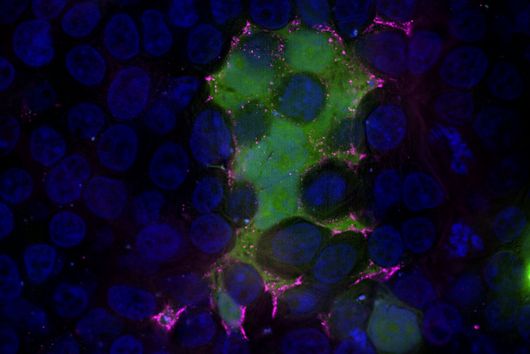
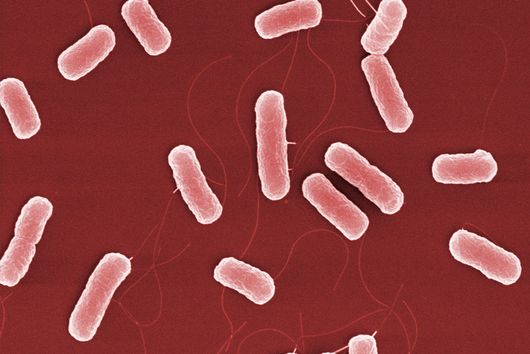
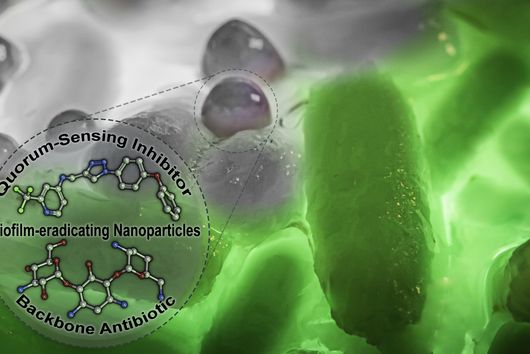
The pathogen Pseudomonas aeruginosa is the cause of a large number of serious infections and places a particular burden on immunocompromised patients. The increasing spread of antimicrobial resistance makes it even more difficult to combat the dreaded hospital pathogen. A research team led by Dr Martin Empting of the Helmholtz Institute for Pharmaceutical Research Saarland (HIPS) has now discovered a new class of active compounds that disrupt the bacterium's chemical communication pathways. This not only reduces the pathogen's disease-causing properties, but also simultaneously enhances the effectiveness of antibiotics. The researchers published their findings in the journal Advanced Science. HIPS is a site of the Helmholtz Centre for Infection Research (HZI) in collaboration with Saarland University.
Although bacteria are among the simplest life forms on our planet, in the course of evolution they have developed ways to communicate efficiently with each other. Unlike humans, their communication is not mediated with words but with chemical signals. This interaction also plays a fundamental role in infection processes of pathogenic bacteria in humans. For example, bacteria communicate to their swarm when it is time to produce substances that counteract the human immune system. Interfering with these communication pathways is a promising starting point for the development of new therapies.
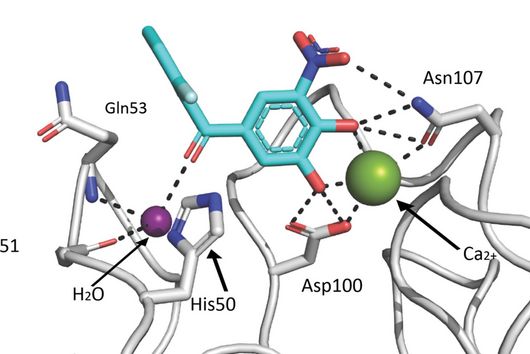
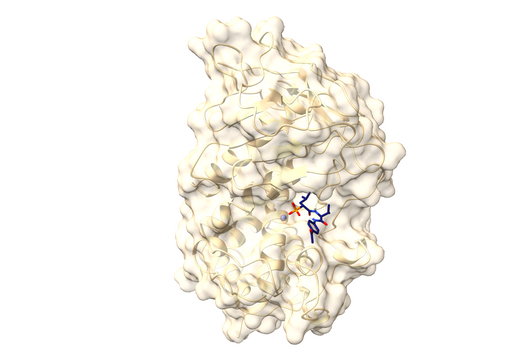
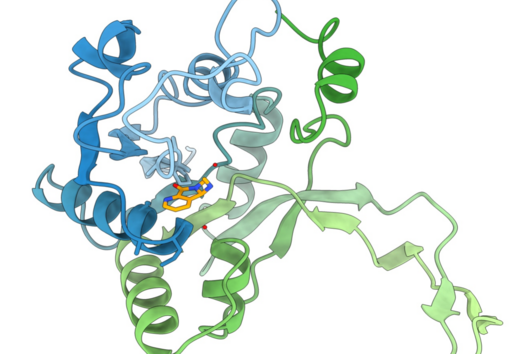
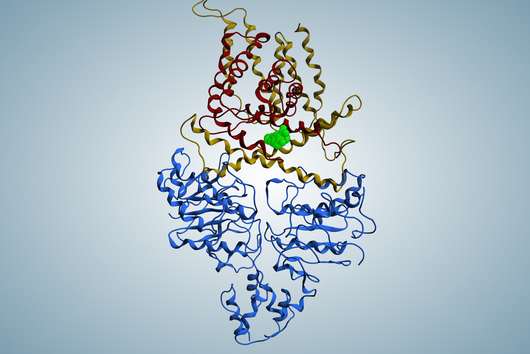
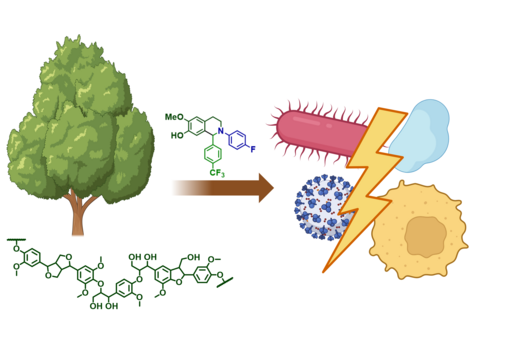
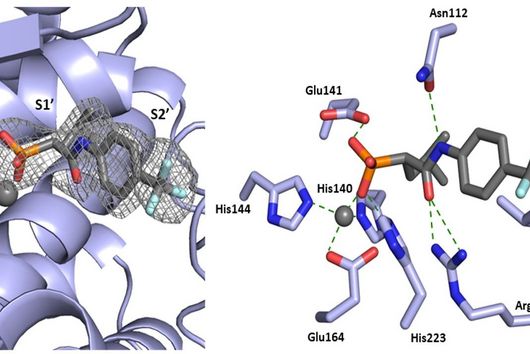
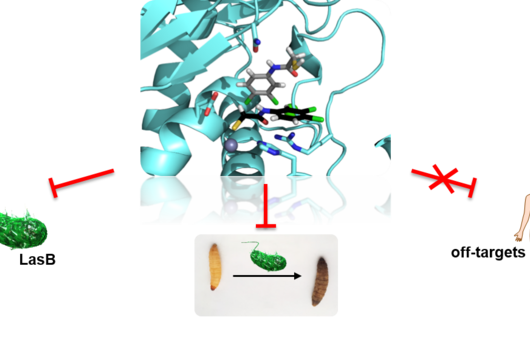
However, the team around Dr Martin Empting, head of the Group Antiviral and Antivirulence Drugs at HIPS, did not focus on the communication messengers themselves, but on their receptors - i.e. the parts of the bacterium that are responsible for signal processing. In this specific case, the researchers targeted the receptor PqsR. When this receptor is activated, inflammation-promoting substances and biofilms are formed, in which P. aeruginosa is largely protected from antibiotics. The drug class now described was chemically designed and optimized by the researchers so that it can attack its target PqsR as efficiently as possible. This was based on structural data obtained by X-ray crystallography.
"Since we always have a potential application in humans as a goal when developing new active substances, we had to achieve not only excellent efficacy but also good pharmacological properties and minimize undesirable side effects," says Martin Empting. He further explains, "This was not always easy, as even small changes to the molecule often have a big impact on its properties, but we can be very satisfied with our result." In further laboratory experiments, the scientists were able to show that the optimized substances reliably prevent the formation of the pro-inflammatory molecule pyocyanin in a large number of clinical isolates of P. aeruginosa. Furthermore, they were able to show that the new active ingredient is capable of impairing biofilm formation and can even further enhance the effect of the antibiotic tobramycin. Finally, the researchers were able to transfer the promising properties of their active substance to a mouse model. Here, the combined administration of tobramycin and the new PqsR inhibitor was able to combat the P. aeruginosa infection significantly better than either substance alone.
The authors see great potential in the new compound class for future application in humans. However, before the substance can be tested in humans, complex preclinical safety studies and development processes still have to be completed. Martin Empting gives the following assessment: "We see the potential for our compounds to be used therapeutically in patients with chronic lung diseases such as cystic fibrosis or bronchiectasis and associated P. aeruginosa infections in a few years' time."
Original publication:
Hamed MH, Abdelsamie AS, Rox K, Schütz C, Kany AM, Röhrig T, Schmelz S, Blankenfeldt W, Arce-Rodriguez A, Borrero-de- Acuña JM, Jahn D, Rademacher J, Ringshausen FC, Cramer N, Tümmler B, Hirsch AKH, Hartmann RW & Empting M: Towards Translation of PqsR Inverse Agonists: From in vitro Efficacy Optimization to in vivo Proof-of-Principle. Advanced Science, 2023, DOI: 10.1002/advs.202204443